To download multiple files we recommend using wget. This is likely to be already installed on Linux,
and is available for Windows and Mac. To download all the trimmed
data files use the command:
This will create a directory "Trimmed" containing all the .fastq.gz files. Be sure to copy
the command exactly and include the trailing "/".
wget -r --cut-dirs=2 -np -nH -R "index.html*" http://cgr.liv.ac.uk/illum/LIMS16056_259a058713a41d9b/Trimmed/
The sequence files are in .fastq.gz format. This is a version of the text FASTQ format, which has been binary compressed using gzip to reduce the file sizes. Many analysis programs such as Velvet and BWA can read .fastq.gz files directly, however for some software packages you may need to decompress the files using gunzip or 7-zip.
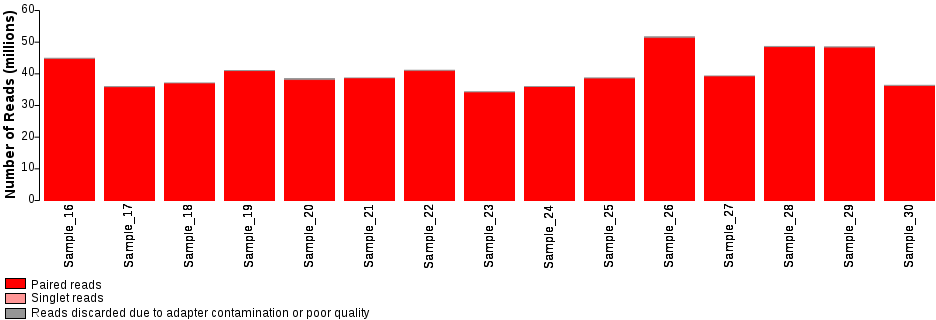
For paired-end sequence data, there are three sequence file types. The files labelled R1 and R2 contain the corresponding paired-end sequences. The singlet files contain sequences whose pair has been removed due to poor sequence quality or adapter contamination. If a sample has been sequenced several times, there will be several sets of sequence files in the sample directory. These will need to be concatenated before downstream analysis.
Diagram illustrating the total number of reads obtained for each sample.
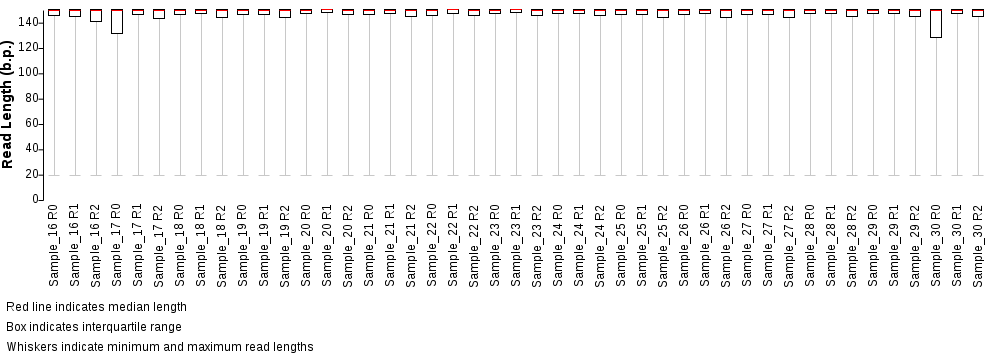
Box plot showing the distribution of
trimmed read lengths for the forward (R1), reverse (R2) and singlet (R0) reads. Note that it is common
for a small number of reads to consist of mostly adapter-derived sequence, so it is expected that
the distribution will show a long tail.
Further detailed statistics are available for each of the trimmed and raw data files.
The raw Fastq files are trimmed for the presence of Illumina adapter sequences using Cutadapt version 1.2.1 [Reference]. The option -O 3 was used, so the 3' end of any reads which match the adapter sequence for 3 bp. or more are trimmed.
The reads are further trimmed using Sickle version 1.200 with a minimum window quality score of 20. Reads shorter than 20 bp. after trimming were removed. If only one of a read pair passed this filter, it is included in the R0 file. The output files from Cutadapt and Sickle are available here.
Statistics were generated using fastq-stats from EAUtils.
Libraries made by Lucille Rainbow; sequencing by Anita Lucaci.
The bioinformatics analysis for this project has been performed by R.Gregory. Please e-mail if you have any further queries.